
Concept Design to Market Launch – The Biorep Advantage
Partner with Biorep on your journey of innovation, where our FDA compliant 5-phase design and development process sets the stage for turning your ideas into reality. In this blog post, we’ll walk you through the key stages of our proven process and illustrate how Biorep can be your trusted partner in bringing your concepts to life by utilizing our 5-Phase Process to generate your Design History File (DHF) and Risk documentation. Our design and development process is more than just a series of steps – it’s a commitment to excellence, aligning seamlessly with ISO 13485 and 21 CFR 820 requirements to ensure that your journey from concept to market launch is marked by quality, compliance, and success.
Compliance – Why do you need the Design and Development process?
The design and development process is a critical component required by both the FDA (Food and Drug Administration) and ISO (International Organization for Standardization) to ensure the safety, efficacy, and quality of medical devices. The FDA, under 21 CFR Part 820, mandates a comprehensive design control system for medical device manufacturers, encompassing procedures and documentation throughout the product development lifecycle. Simultaneously, ISO standards, particularly ISO 13485, emphasize the necessity of a robust design and development process, reinforcing global best practices for quality management systems in the medical device industry. Adhering to these stringent requirements not only facilitates regulatory compliance but also fosters innovation with a focus on risk management, usability, and traceability – essential elements for bringing safe and effective medical products to market. The synergy between FDA and ISO standards in the design and development process underscores the commitment to ensuring that medical devices meet the highest standards of quality and regulatory compliance.
Certain medical devices are exempt from specific design control requirements according to FDA regulations and ISO standards. Our team will help determine the class of your device and will help develop a regulatory plan catered to the needs of your device.
The FDA also requires the creation of a Design History File (DHF) and Risk Management documentation which is outlined in 21 CFR 820.30, a crucial component of the Quality System Regulation (QSR) for medical devices. By maintaining a DHF, manufacturers can effectively manage risks associated with design changes, traceability issues, and regulatory compliance throughout the product lifecycle, thereby enhancing patient safety and product effectiveness while facilitating regulatory oversight and quality assurance. Biorep has established procedures for identifying, assessing, and mitigating risks associated with your device throughout its product lifecycle, starting with the Design and Development process. Compliance with this requirement ensures that potential hazards are proactively addressed, contributing to the safety and effectiveness of medical devices on the market. Biorep will develop a DHF and Risk documentation for you that will withstand the most rigorous regulatory audit.
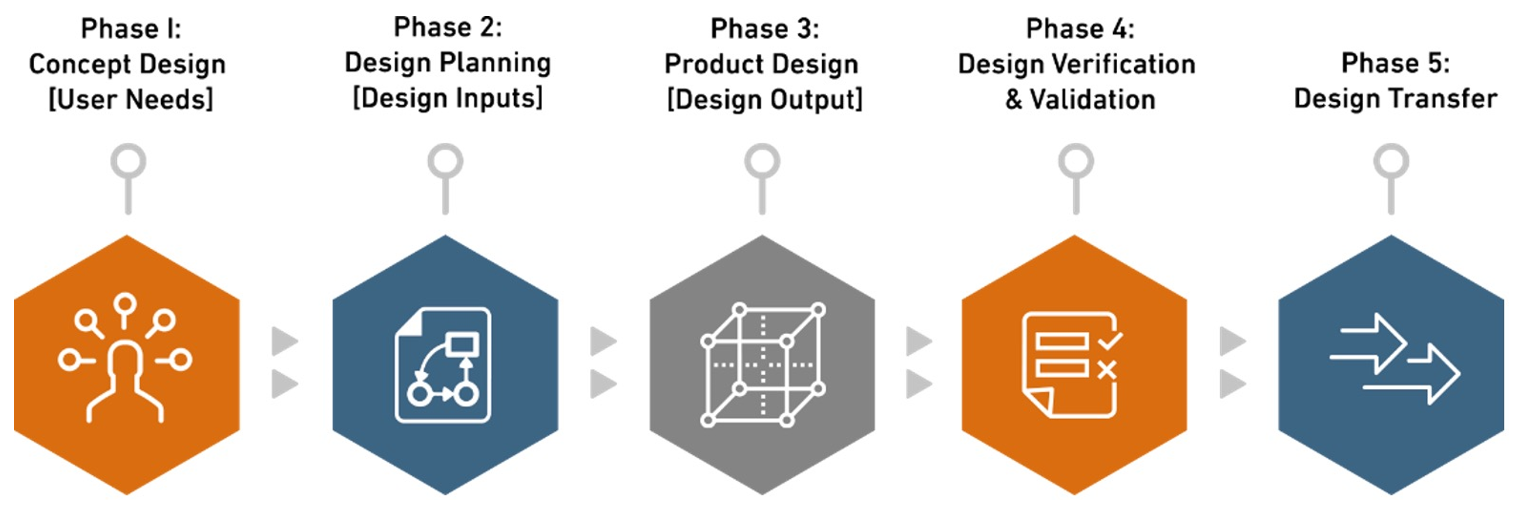
Our 5 Phased Approach
Phase 1. Concept Design [User Needs]
User needs in design and development for medical devices encompass the specific requirements and expectations of end-users or customers, fundamentally shaping functionality, performance, usability, safety, and regulatory compliance. Identified early in the development phase, these needs define the device’s purpose, features, accuracy, efficiency, user interface, and safety standards, guiding subsequent design activities. During the concept design phase, Biorep’s Product Development team collaborates closely with clients to comprehensively define these user needs, leveraging expertise to anticipate challenges and mitigate risks from the beginning. By understanding key requirements and end-users at the project’s onset, Biorep ensures a solid foundation for a successful design process, focusing not only on creating a product, but crafting a device that resonates with the target audience.
Phase 2. Design Planning [Design Inputs]
Design inputs serve as the foundational elements defining the physical and performance characteristics of a medical device, essential for translating user needs into specific engineering requirements and ensuring regulatory compliance. Providing a comprehensive set of specifications, design inputs guide the development team throughout the design process, facilitating the creation of a product that meets user expectations and industry standards. Biorep’s engineers conduct in-depth research regarding critical parameter and translate user needs into engineering requirements (Design Inputs) that align with clients’ and users’ vision.
Phase 3. Product Design [Design Output]
Design outputs, including specifications, drawings, and manufacturing instructions, outline the physical and performance characteristics of a medical device, ensuring alignment with defined design inputs. These outputs are pivotal in establishing device conformance, safety, and functionality, addressing elements such as performance, manufacturing instructions, quality criteria, packaging, and traceability. Integral to the design control process, they provide guidance for manufacturing and Quality Assurance, facilitating Verification and Validation to meet design criteria and user needs effectively. In this phase, conceptual ideas materialize as Biorep’ s team establishes design outputs which are the basis for design verification methods, striving for excellence in functionality, aesthetics, and user experience. Here, products transition from concepts to tangible prototypes, ready for rigorous testing, with our goal being a seamless transition from concept to design.

Phase 4. Design Verification & Validation
In the design and development process of medical devices, Design Verification (DV) and Validation (DVAL) are pivotal stages ensuring the product meets specified requirements and functions as intended. Design Verification involves systematic review, inspection, or testing to confirm established design outputs meet design inputs, ensuring thorough scrutiny of each aspect. Conversely, Design Validation assesses whether the device satisfies user needs, conducted on the final product considering the entire manufacturing process. Real-world testing and evaluation simulate actual usage conditions, ensuring compliance with intended use requirements and user expectations.
Comprehensive documentation supports regulatory compliance, becoming part of the Design History File (DHF), crucial for submissions. Both DV and DVAL activities are conducted following established protocols and standards, reinforcing the device’s safety, efficacy, and quality. At Biorep, quality is the cornerstone of our process. In the design verification and validation phase, we rigorously test and assess the product against design inputs and user needs, surpassing standards to ensure exceptional compliance and user satisfaction. Our rigorous testing and unwavering commitment to Quality ensures a device that exceeds expectations.
Phase 5. Design Transfer
Design Transfer is a pivotal phase in the design and development process, marking the transition from the finalized device design to commercial manufacturing. This meticulous process involves translating detailed device designs into practical manufacturing instructions and specifications, aiming for a seamless transition to ensure replication of the design intent during production. It encompasses releasing development procedures, instructions, and specifications to the production team, facilitating the transformation of conceptualized designs into tangible, reproducible products. Throughout Design Transfer, equipment and process validations are conducted to ensure alignment with established design outputs, with documented protocols, results, and conclusions essential for verifying manufacturing processes’ repeatability and reliability amidst day-to-day variables.
The collaborative efforts of Engineering, Manufacturing, Quality, and Regulatory teams are crucial for approving these validations, ensuring a smooth transition compliant with regulatory standards. Design Transfer represents the final phase before product launch, indicating the device’s readiness for commercialization. As the design and development journey culminates, Biorep ensures a seamless transition to commercial manufacturing and the marketplace. With a focus on maintaining the highest standards of quality, we handle every aspect, from legal agreements to supplier qualifications, providing assurance that your product is primed for success in the competitive landscape.
Taking the Next Step on your Product Development Journey
Every project is unique, and we understand that. Putting the time and effort into the Product Development process from the beginning and partnering with the right design and development company will save time, money and reduce any future regulatory burdens.
Contact us today to learn more about our solutions and if they are the right fit for your medical device.